

第一作者:Zhou Wang
通讯作者:刘继磊 教授
单位:湖南大学
https://doi.org/10.1002/aenm.202202874
【背景】
粘结剂是电极中的一个重要组成部分,除了保持电极的结构、机械和电气完整性外,它在形成稳定的SEI层方面也发挥了重要作用。事实证明,包括羧甲基纤维素钠(CMCNa)和聚丙烯酸钠(PANa)在内的水溶性粘结剂在缓解电解质分解和提高石墨电极的K+存储性能方面比通常使用的聚偏氟乙烯(PVDF)对应物更有优势。与PVDF系统相比,CMCNa和PANa的初始效率也得到了改善。据报道,在锂离子电池中,聚丙烯酸酯(PAA)粘结剂促进了LiFSI的快速分解,导致了富含LiF的SEI层的形成,而钝化硫化物的分布是均匀的。与羧甲基纤维素钠(CMC)系统相比,这使得PAA粘结剂系统具有更好的容量保持能力。所有这些都突出了粘结剂在物理、化学和机械方面的重要贡献。然而,粘结剂在调节电解质还原反应和机制、SEI形成率及其成分分布等界面化学方面的作用还没有得到很好的理解,阻碍了合理设计粘结剂以进一步提高PIB性能的研究工作。
【工作介绍】
本工作介绍了基于羧甲基纤维素钠(CMC)和基于聚偏氟乙烯(PVDF)的石墨电极作为模型系统,以量化电解质分解、固体电解质界面(SEI)的形成以及相应的动力学演变转变。在还原过电位和产物选择性方面,确定了基于CMC的电极上的非催化性电解质还原路径和基于PVDF的电极上的催化性还原路径。电解液还原和/或SEI的形成被发现是以连续的两步方式发生的,首先是在0.35V以上的电位下进行电化学还原,在0.35V以下的电位下进行热力学加速的电解液还原(步骤II)。动力学分析表明,前者对于基于CMC和PVDF的电极都是电荷转移控制的,后者涉及到PVDF系统的动力学过渡到SEI电阻控制,而对于CMC系统是电荷转移控制的。所有这些例子,强调了粘结剂化学在电解质分解和电极/电解质界面特性中起着主导作用,并促进了对电解质还原的更好的基本理解。
【具体内容】
石墨电极:85wt.%的天然石墨、5wt.%的科琴黑和10wt.%的干粘合剂(PVDF或CMC或SA);K金属作为对电极;电解液:0.8M KPF6 in EC and DEC(v/v = 1:1)。
电化学性能的比较
图1a,b表明了基于CMC和PVDF的石墨电极的恒流充电-放电(GDC)曲线和相应的差分容量与电位(dQ/dV)曲线。对于基于CMC的石墨电极,除了与低于0.35V的K+插层到石墨中有关的阶段性特征和在≈0.32V对应于K+提取的阳极峰外,在≈0.50V也发现了额外的阴极峰,但在随后的循环中没有。这被认为是形成了一个稳定的钝化层,保护表面免受进一步的电解质还原。对于基于PVDF的电极,在放电曲线中低于0.40V的类似阶段性特征以及在≈0.33V左右的去势特征已经被确定。但是对于PVDF基电极来说,初始循环中的不可逆还原反应是在≈1.0V的正电位下开始的,而CMC基电极则是在≈0.80V左右。此外,PVDF基电极的初始库仑效率(ICE)估计为39.8%,远低于CMC基电极的效率(73.9%)。这表明相对于CMC基电极而言,PVDF基电极涉及更严重的电解质还原。
根据dQ/dV分析,初始放电曲线可以分为两个区域,一个是电位范围从OCV到大约0.35V(区域I,没有发生嵌钾),另一个是电位低于0.35V(区域II)。对于PVDF基的电极,大约49.87%的不可逆容量损失发生在区域I,这比CMC基的电极(4.47%)高得多。这意味着:i)即使在很正的电位范围内(在区域I),PVDF系统也会出现严重的电解质还原;ii)区域II(在钾化石墨电极上)的电解质还原对于PVDF和CMC系统都是主要的。因此,有理由推测,电解质还原和/或SEI的形成由两个步骤组成,即区域I的温和反应和区域II的加速还原。考虑到在区域I中还没有嵌钾,步骤I被指定为电化学反应,而步骤II与区域II中钾化石墨上热力学加速的电解质还原有关。这意味着电解质还原以连续的、两步的方式发生。
所有系统的CE都随着循环数的增加而明显增加,但遵循同样的趋势,即CMC>PVDF。这些差异还表现在循环稳定性和速率能力上,基于CMC的电极表现出卓越的K+储存行为,以上表明石墨中K+储存的强烈粘结剂化学依赖性质。而对于SA(海藻酸钠),提供了与CMC系统相当的电化学性能,并优于PVDF系统。鉴于相同的实验条件,不同的电解质还原过电位和/或程度,以及不同的K+存储行为(即CE,不同电位范围内的不可逆性,循环性能等),揭示了i)每个电极上的不同反应,以及ii)水性粘结剂优于PVDF。
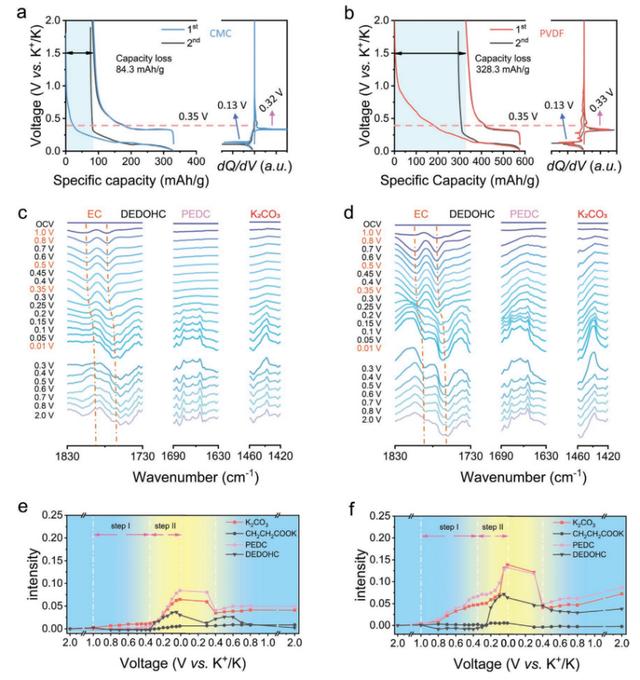
图1-电化学性能和还原活性的比较。a)CMC基和b)PVDF基电极的静电充电-放电曲线和相应的dQ/dV曲线。c,d)原位ATR-FTIR光谱和e,f)还原产物的相应峰强演变。c,e)CMC基和d,f)PVDF基电极。
电解质还原路径的表面化学依赖性
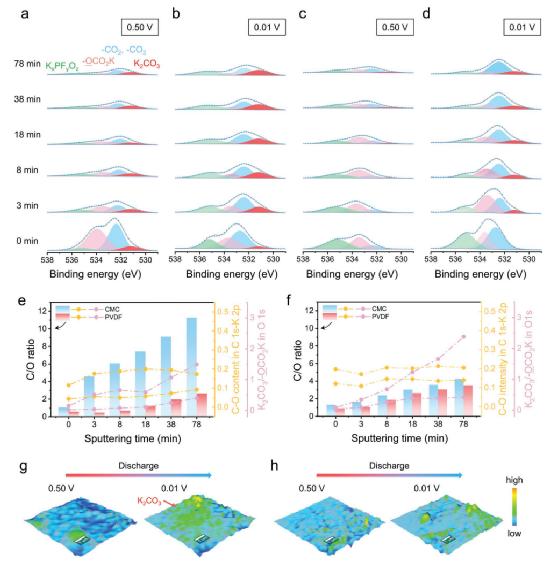
图2–a,b,e,g)CMC衍生的SEI和c,d,f,h)PVDF衍生的SEI的特征。a,b)CMC衍生的SEI和c,d)PVDF衍生的SEI的O 1s光谱比较,在放电电位为0.5和0.01V时,不同的Ar+轰击时间。e,f) 定量分析C/O的比例,C 1s-K 2p中C-O的含量,以及O 1s中K2CO3 /-OCO2K。g,h) 在0.5和0.01 V的放电电位下收集K2CO3物种的拉曼映射。
电解质反应机制
状态密度(DOS)计算结果表明,EC(或DEC)在电解质的最低未占分子轨道(LUMO)能量值中占主导地位(图3a),因此EC更倾向于被还原并参与石墨电极上的SEI形成。提出了涉及竞争反应(反应I和II)和相互转化(反应III)的方案(图3b),以说明CMC和PVDF基电极上表面化学依赖的过电位和还原选择性。其中,K2CO3是由EC的直接双电子还原(反应I)或由ROCO2K与微量水的二次反应(反应III)形成的。EC的分子还原电位≈0.9V vs. K/K+,这取决于其溶剂化状态。基于CMC的电极上的电解质还原电位约为≈0.8 V,于EC的理论值(≈0.9 V),表明溶剂在基于CMC的石墨电极表面的还原活性被抑制。在基于CMC的电极上首先触发了反应I(≈0.8 V),然后是反应II(≈0.5 V)。
反应I和反应II在进一步放电时相互竞争,这一点从FTIR结果中可以看出(图1e)。这也很好地解释了以下事实:i)在步骤I中形成更多的K2CO3,以及ii)在步骤II中K2CO3和PEDC的积累是相当的。相反,基于PVDF的电极上的还原反应在一个更高的电位(≈1.0V)开始(图1f),表明出一个典型的催化还原过程。这可以归因于基于PVDF的电极表面与溶剂分子更强的相互作用,降低了后续电子转移步骤的障碍,促进了EC的还原活性,以及PVDF更差的电化学稳定性(由更高的起始还原电位和更大的限制电流证明)。
此外,基于PVDF的电极的还原选择性与基于CMC的电极不同。在基于PVDF的石墨电极表面,包括K2CO3和PEDC的还原产物在≈1.0 V的电位下同时发生,并且在步骤I中检测到更多的PEDC(图3e)。这表明反应II在这里首先被触发,同时伴随着反应III。不能排除反应I(在步骤I中)的存在,尽管缺乏直接的证据。尽管如此,鉴于K2CO3(图1e,f)的峰值强度急剧增加,反应I无疑是在步骤II中触发的。但总的来说,相对于基于CMC的电极,在基于PVDF的电极上产生的K2CO3更少(图2e,f)。所有这些差异意味着反应I在CMC系统中更有利,而反应II在PVDF系统中热力学上更有利。
图3-a) 电解质的状态密度(DOS)。 b)电解质还原途径,包括竞争反应(反应I和II)和相互转化(反应III)。c) 总结的反应I和II的吉布斯自由能。
对于EC分子来说,有两种可能的开环反应路径(过渡态O2 -C-O-CH2 -CH2 vs O-C-O-CH2 -CH2 -O),导致不同的中间产物和最终产物。DFT计算表明,尽管CMC-EC+e和PVDF-EC+e分子之间的能量屏障有很大不同,但反应路径I对所有粘结剂系统都是能量上有利的(图3)。对于反应路径I,涉及两个中间产物M1和M2,能量屏障按CMC-和PVDF-分子的顺序递减。这进一步证实了PVDF基电极上更高的EC反应活性。M2-K是由M2与钾离子结合形成的,这个分子有两种可能的反应途径(图3d)。一种途径是M2-K捕获一个电子和一个K+,形成K2CO3。另一条途径涉及对M2-K复合体的核攻击,导致PEDC的形成。这两个反应在热力学上都是有利的,但在吉布斯自由能(ΔG)方面有所不同。对于CMC系统,形成K2CO3和PEDC的总ΔG为-575.2和-363.8KJ mol?1,ΔG差为-210KJ mol?1(图3c)。所有这些数值都比PVDF系统的数值(-534.6和-330.8KJ mol?1)低很多。这表明,与反应II相比,反应I更可取,特别是对于基于CMC的电极。
进一步进行了AIMD模拟。其中,乙烯碳(Ce)和乙烯氧(Oe)的局部状态密度(LDOS)提供了一个明显的负迁移,说明在水基粘结剂和PVDF系统中,EC分子的热力学上是有利的减少。具体来说,SA/CMC体系中Ce -Oe两边的LDOS曲线是对称的,表明EC在这里容易发生Ce-Oe键裂解(通过反应I),形成更稳定的CO32?阴离子。这也被拉长的Ce -Oe键的长度(分别为1.47和1.49?)所验证。相比之下,PVDF系统的DOS和键长曲线都是不对称的,表明Ce -Oe键的单侧断裂(反应II)产生了PDEC。这很好地解释了原位FTIR结果和深度剖面XPS分析的结果,即反应I对于基于CMC和基于SA的电极是主要的,而基于PVDF的电极在热力学上对反应II更有利。因此,根据DFT计算、AIMD模拟和原位ATR FTIR分析,合理的结论是,对于基于PVDF的电极来说,是反应III促成了K2CO3,而不是反应II。反应I直接产生丰富的K2CO3,没有进一步的分解,有利于形成一个紧凑和高度稳定的SEI,因为参与的气体产物较少。相反,反应II产生丰富的PEDC,它几乎是一个离子绝缘体,电化学不稳定,在循环时分解成更多的气体产物,如RCH=CH2,O2,和CO2,和/或通过反应III立即转变为K2CO3。这转化为每个电极上不同的钝化层特性(图3e)。具体来说,SEI物种的峰值强度在充电时逐渐下降(图1c-h),但CMC衍生的SEI的整体物种变化(即K2CO3、PEO、PEDC等)相对于PVDF衍生的SEI来说要小得多,这进一步证明了CMC在形成完整和高度稳定的SEI层方面的优势。所有这些,都很好地解释了基于CMC的电极比基于PVDF的电极具有更好的K+储存行为。
总之,电解质还原和/或SEI的形成是以一种连续的、两步的方式发生的,并带来强烈的表面化学依赖性。与第一步中温和的电化学反应相比,第二步中热力学加速的电解质还原在钾化石墨上被高度促进。此外,主导整个电解质分解过程的是后者,而不是前者。然而,差异在于还原的选择性、程度和过电位,这源于CMC和PVDF之间固有的化学差异。具体来说,步骤I中的电解质还原在基于CMC的电极上被明显抑制,可能是得益于水基粘结剂中的羧酸钠与HF反应,从而抑制了溶剂和盐的还原。反应I在CMC系统中占主导地位,而反应III在PVDF系统中更为有利。
动力学转换
通过采用原位EIS与松弛时间分布(DRT)分析相结合的方法,揭开了从K动力学演变中得出的电化学特征的巨大差异。典型的EIS(在一个三电极装置中收集)是由高/中高频的两个重叠的半圆和低频端的一条直线(10KHz至0.1 Hz)组成的,它们的轮廓随着电位的变化而变化(图4a,d)。高频下不完整的半圆归因于在电极表面形成的SEI层,电阻被表示为RSEI。Rct是法拉第的电荷转移电阻,对应于中频下的半圆。最初放电时的阻抗演变也分为两个阶段,由两步电解质还原过程组成(图4a,d)。在第一步中,半圆随着电压的降低而不断扩大。这归因于初步的SEI的形成,它具有很强的抗性。当K+插到石墨中时,半圆表现出持续的收缩,直到放电结束,这意味着高导电的SEI在步骤II中逐渐形成。尽管基于CMC和PVDF的电极都有类似的趋势,但CMC衍生的SEI的RSEI要比PVDF系统的小,这得益于其薄、完整和坚固的特点。
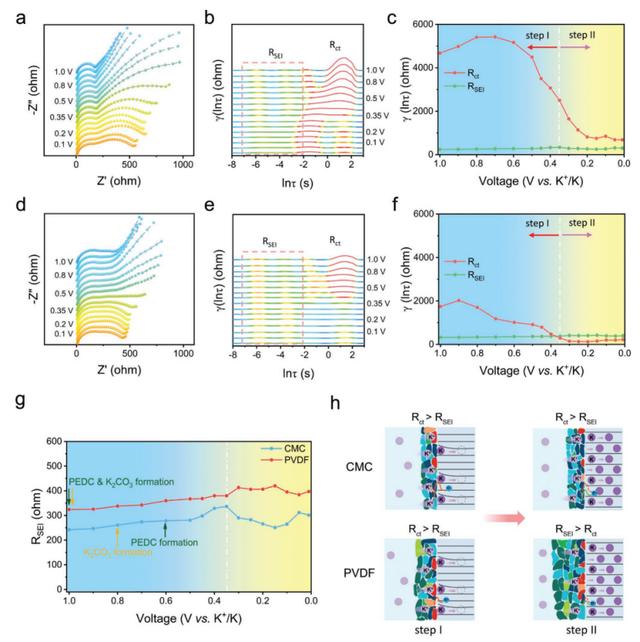
图4-电解液还原和/或SEI形成后的动力学演变。a) 基于CMC的电极和d) 基于PVDF的电极的原位EIS。b,e) 相应的计算DRTs和c,f) Rct , RSEI的计算阻抗结果。g) 比较RSEI的演变。h) 连续的两步电解质还原过程的示意图。
为了精确区分特定的电化学过程,松弛时间分布(DRT)方法被用来在松弛时间的空间中对阻抗数据进行分解。从本质上讲,它是一个纯粹的数学转换,通过公式(1)将频域Nyquist图转化为时域DRT曲线。

其中,每个物理过程都被表示为连续分布函数的局部最大值。而峰下的面积代表阻抗值。对于基于CMC和基于PVDF的石墨电极,在lnτ范围内的-8到3s,电化学过程的两个特征峰(τ1 , τ2)被清楚地确定(图4b,e)。位于-8至-2s的峰值归因于界面反应,即固体电解质界面的形成和K+去溶剂化以及随后通过SEI层的迁移(阻抗表示为RSEI)。它对总极化的贡献很小。位于-2至3秒的松弛过程归因于石墨电极的电荷转移反应(阻抗表示为Rct)。在步骤I中,RSEI持续增加,意味着连续形成的SEI层是电阻性的(图4b-g)。差分容量-电压图(dQ/dV,图1a)表明,在这个步骤中,有非常少的电荷参与。因此,我们推测在步骤I中SEI膜的生长很可能来自于溶剂和阳极之间的化学反应。
形成的SEI可能是一个简单的吸附层,提供高电阻。而且CMC衍生的SEI的RSEI值比PVDF衍生的SEI小,突出了CMC粘结剂在形成高导电性的SEI方面的优势。虽然Rct在初始放电时略有增加,然后急剧下降,直到电位达到≈0.35 V(步骤I的结束)。但是Rct在这里仍然主导着CMC和PVDF系统的整个电化学过程(Rct >> RSEI)。这是可以理解的,因为在SEI-阳极界面上没有发生钾离子的电化学过程,导致Rct值非常高。具体来说,与基于CMC的电极相比,基于PVDF的电极的Rct值较小。这归因于:
i)PVDF固有的较高的电子和离子导电性;
ii)PVDF通过吸收更多的电解质促进膨胀和/或渗透的特点。所有这些都有利于基于PVDF的电极中的电荷转移。
当进入第二步时,上述趋势变得不同了,在这一步中,钾化过程开始(在≈0.35V)。对于PVDF衍生的SEI,RSEI持续增加,同时Rct急剧下降。对于PVDF基电极上的电解质还原,从Rct控制(在步骤I)到RSEI控制(在步骤II)的动力学过渡被解读出来(RSEI > Rct)。然而,在基于CMC的电极上,电解质还原(在步骤I和II)是Rct占主导地位。这些差异与SEI薄膜的纹理结构和成分的重建有关,也与活性电极的体积变化有关。具体来说,在PVDF系统中,PVDF基电极的高反应性,其中涉及更严重的电解质还原(图1d,h),电子/离子绝缘的PEDC的积累。和引起大量体积变化的膨胀效应,说明了RSEI到Rct的过度。相反,高导电性的SEI膜在基于CMC的电极上形成,受益于其PEO和K2CO3 -丰富的特征和完整的薄的特性。
【结论】
总之,粘结剂化学在电解质分解过程、SEI形成和成分/动力学演变中的主导作用被清楚地解析出来。揭示了基于CMC的电极上的非催化还原路径和基于PVDF的电极上的催化还原路径。并确定了一个连续的、两步的电解质还原方式。
研究发现,SEI的形成包括在0.35V以上的电位下的电化学还原(步骤I)和热力学加速电解质在钾化石墨上的还原(步骤II)。热力学加速的电解质还原在整个电解质分解过程中占主导地位。所有这些都有助于形成不同的电极/电解质界面特性。在基于CMC的石墨电极上形成了一个薄的、完整的、坚固的SEI层,其中富含PEO和K2CO3,而富含PEDC的PVDF衍生的SEI则很厚且易碎。所有这些都转化为CMC基电极比PVDF基电极更优越的K+储存性能。这些结果促进了对粘结剂化学对电解质还原的依赖性的更好的基本理解,并为更好的钾离子电池的合理粘结剂设计打开了新思路。
Binder Chemistry Dependent Electrolyte Reduction in Potassium-Ion Batteries: A Successive, Two-Step Reduction WayAdvanced Energy Materials ( IF 29.698 ) Pub Date : 2022-11-22 , DOI: 10.1002/aenm.202202874
Wang Zhou, Bingchen He, Lijiao Quan, Ruhong Li, Yuqing Chen, Changling Fan, Shi Chen, Chaohe Xu, Xiulin Fan, Lidan Xing, Jilei Liu
创业项目群,学习操作 18个小项目,添加 微信:923199819 备注:小项目!
如若转载,请注明出处:https://www.zodoho.com/70191.html
相关推荐
-
未来发展十大前景行业,未来发展最有潜力的行业
随着社会经济的发展,一些行业发展迅速。人们都希望找到一份高薪且有前途的工作,那么你知道未来哪些行业会发展得更好吗?下面的第一个列表介绍了未来十大有前途的行业。让我们看看。未来发展十…
-
以服务为本 提高护理服务品质
——专访商丘市中心医院护理部主任王丽 商丘市中心医院护理部主任王丽 □东方今报·猛犸新闻记者 陈涛 ■见习记者 邵显哲 从青春年少到渐生华发,业务从稚嫩到成熟到老练再到炉火纯青,为…
-
豆瓣评分9.0以上的悬疑电影国产,豆瓣评分9.0以上的悬疑电影国产剧
今天给姐妹们安利几部超高水平的欧美悬疑片,每一部都可称深,绝了!既有烧脑神剧,也有黑色幽默喜剧,更有重口味带惊悚元素的悬疑片。 喜欢看欧美悬疑剧的集美,建议收藏。 《终局》 这是今…
-
创业180天,我发现了工具型SaaS的最佳实践
产品的切入点不同,所带来的结果也会有所不同。三句话介绍产品,听得下去的人成交率80%,听不下去的多难以继续合作。本文以该创业公司的故事为例,聊聊工具型SaaS PMF时会遇到的问题…
-
傻子瓜子加盟费多少,零食加盟店10大品牌前三名
节后返沪开工第一天,来自芜湖的大微突然发现自己给办公室同事,准备的老家特产“傻子瓜子”竟然落在老家了。 于是他只能一边夸赞其他同事分享的老家特产好吃,一边尴尬de卖关子笑称,自己的…
-
干洗店的前景怎么样?
干洗店相对其他行业,投资是比较小的;现在哪怕是一个奶茶店,看起来只需有几千块的机器加一些食材,可奶茶店的选址必须在人流量特别大的地方,这样的地方房租都是寸土寸金的,动辄一二十万的转…
-
苏马荡楼盘价格,苏马荡2020年避暑情况
小说《盛夏的云朵》故事梗概及说明 小说《盛夏的云朵》讲述了2017年底万利高速公路通车后,谭先礼一家人及周围朋友们去来利川市谋道镇苏马荡避暑康养度假游玩的寻常故事,反映苏马荡这个最…
-
定期到期后不取怎么算,定期存款到期不取会作废吗
在疫情的这几年,越来越多的人感受到存钱的重要性,因为不知道什么时候会发生意外,存一定的钱,在意外发生的时候能给自己留一个保障。 存的钱多了,很多人都会把这些钱拿去银行做定期存款,因…
-
3万元起步,选择在农村自主创业,现在可以选择哪些适合的项目?
当下,随着互联网普及和新农村的建设,农村的创业项目有很多,有一些都是不需要大投入的。回答这个粉丝的问题,三万元起步,选择在农村自主创业,现在可以选择哪些适合的项目?我这里推荐一些,…
-
2020不打工了,这10个最赚钱的小生意,一个人做月入可能2万以上
说起小吃赚钱项目,大家想到的肯定是烤串、麻辣烫、章鱼小丸子之类的,这些小吃的美味自然就不用多说了,今天我带给大家的就是2019新型小吃最赚钱的好生意,对消费者来说,不仅美味价格还很…
